Kā atpazīt Rett sindromu

Saturs
- Reta sindroma iezīmes
- Kā tiek noteikta diagnoze
- Dzīves ilgums
- Kas izraisa Rett sindromu
- Rett sindroma ārstēšana
Reta sindroms, ko dēvē arī par smadzeņu atrofisku hiperamonēmiju, ir reta ģenētiska slimība, kas ietekmē nervu sistēmu un skar gandrīz tikai meitenes.
Bērni ar Reta sindromu pārstāj spēlēt, kļūst izolēti un zaudē iemācītās prasmes, piemēram, staigāšanu, sarunu vai pat roku kustināšanu, izraisot piespiedu roku kustības, kas raksturīgas slimībai.
Reta sindromu nevar izārstēt, bet to var kontrolēt, lietojot zāles, kas, piemēram, mazina epilepsijas lēkmes, spazmas un elpošanu. Bet fizioterapija un psihomotorā stimulācija ir ļoti noderīga, un vēlams, lai tā būtu katru dienu.
Reta sindroma iezīmes
Neskatoties uz simptomiem, uz kuriem visbiežāk tiek pievērsta vecāku uzmanība, parādās tikai pēc 6 dzīves mēnešiem, mazulim ar Reta sindromu ir hipotonija, un vecāki un ģimene to var redzēt kā ļoti “labu” bērnu un viegli kopjamu.
Šis sindroms attīstās 4 fāzēs, un dažreiz diagnoze tiek sasniegta tikai aptuveni 1 gada vecumā vai vēlāk, atkarībā no pazīmēm, kuras katram bērnam ir.
Pirmais posms, notiek no 6 līdz 18 dzīves mēnešiem, un ir:
- Bērna attīstības apturēšana;
- Galvas apkārtmērs neatbilst normālai augšanas līknei;
- Samazināta interese par citiem cilvēkiem vai bērniem, ar tieksmi norobežoties.
Otrais līmenis, notiek no 3 gadu vecuma un var ilgt vairākas nedēļas vai mēnešus:
- Bērns daudz raud pat bez redzama iemesla;
- Bērns vienmēr paliek aizkaitināts;
- Parādās atkārtotas roku kustības;
- Parādās elpošanas izmaiņas, dienas laikā apstājoties elpošanai, palielināta elpošanas ātruma brīdis;
- Konvulsīvi krampji un epilepsijas lēkmes visas dienas garumā;
- Miega traucējumi var būt bieži;
- Bērns, kurš jau runāja, var pilnībā pārtraukt runāt.


Trešā fāze, kas notiek apmēram pirms 2 un 10 gadiem:
- Līdz šim parādītie simptomi var nedaudz uzlaboties, un bērns var atgriezties, lai izrādītu interesi par citiem;
- Bagāžnieka pārvietošanas grūtības ir acīmredzamas, ir grūtības piecelties;
- Var būt spastiskums;
- Attīstās skolioze, kas pasliktina plaušu darbību;
- Miega laikā ir ierasts griezt zobus;
- Barošana var būt normāla, un arī bērna svars mēdz būt normāls, nedaudz palielinoties svaram;
- Bērnam var pazust elpa, norīt gaisu un pārāk daudz siekalu.
Ceturtā fāze, kas notiek apmēram 10 gadu vecumā:
- Kustības zaudēšana pamazām un skoliozes pasliktināšanās;
- Garīgais deficīts kļūst smags;
- Bērni, kuri spēja staigāt, zaudē šo spēju un viņiem nepieciešams ratiņkrēsls.
Bērniem, kuri var iemācīties staigāt, joprojām ir zināmas grūtības pārvietoties, un viņi parasti pirksto vai sper pirmos soļus atpakaļ. Turklāt viņi, iespējams, nespēj nekur nokļūt, un viņu pastaiga, šķiet, ir bezmērķīga, jo viņa nestaigā, lai satiktu citu cilvēku vai, piemēram, paņemtu kādas rotaļlietas.
Kā tiek noteikta diagnoze
Diagnozi nosaka neiropediatrs, kurš detalizēti analizēs katru bērnu atbilstoši norādītajām pazīmēm. Diagnozei jāievēro vismaz šādas īpašības:
- Acīmredzot normāla attīstība līdz 5 dzīves mēnešiem;
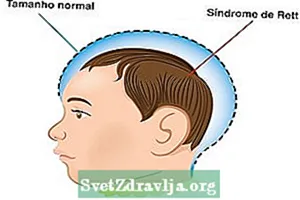
- Normāls galvas izmērs piedzimstot, bet pēc 5 dzīves mēnešiem tas neatbilst ideālajam mērījumam;
- Spēju normāli pārvietoties ap 24 un 30 mēnešiem zudums, kas izraisa nekontrolētas kustības, piemēram, pagriežot vai pieliekot rokas pie mutes;
- Šo simptomu sākumā bērns pārtrauc mijiedarboties ar citiem cilvēkiem;
- Bagāžas kustību un nekoordinētas staigāšanas koordinācijas trūkums;
- Bērns nerunā, nevar izpausties, kad kaut ko vēlas, un nesaprot, kad mēs ar viņu runājam;
- Smaga attīstības aizture, sēžot, rāpojot, runājot un ejot daudz vēlāk, nekā paredzēts.
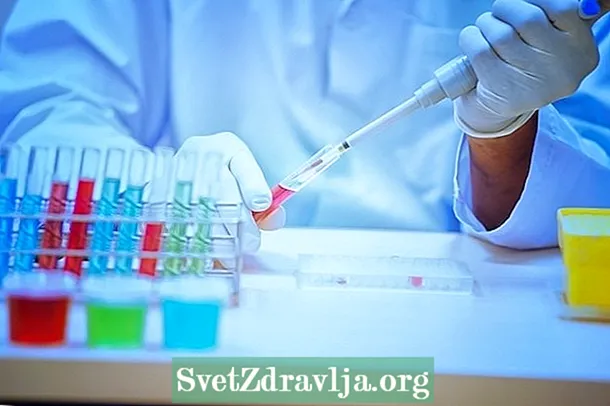
Vēl viens uzticamāks veids, kā uzzināt, vai šis sindroms patiešām ir, ir veikt ģenētisko testu, jo aptuveni 80% bērnu ar klasisko Rett sindromu ir MECP2 gēna mutācijas. Šo pārbaudi SUS nevar veikt, taču privātie veselības plāni to nevar noliegt, un, ja tas notiek, jums jāiesniedz prasība tiesā.
Dzīves ilgums
Bērni, kuriem diagnosticēts Reta sindroms, var dzīvot ilgu laiku, pārsniedzot 35 gadu vecumu, bet gulēšanas laikā var piedzīvot pēkšņu nāvi, kamēr viņi vēl ir bērni. Dažas situācijas, kas veicina nopietnas komplikācijas, kas var izraisīt letālu iznākumu, ir infekciju klātbūtne, elpceļu slimības, kas attīstās skoliozes un sliktas plaušu paplašināšanās dēļ.
Bērns var apmeklēt skolu un var mācīties noteiktas lietas, taču ideālā gadījumā tas būtu jāintegrē speciālajā izglītībā, kur tā klātbūtne nepievērsīs lielu uzmanību, kas varētu pasliktināt mijiedarbību ar citiem.
Kas izraisa Rett sindromu
Reta sindroms ir ģenētiska slimība, un parasti skartie bērni ir vienīgie ģimenē, ja vien viņiem nav dvīņu brāļa, kuram, iespējams, būs tāda pati slimība. Šī slimība nav saistīta ar vecāku veiktu darbību, un tāpēc vecākiem nav jājūtas vainīgiem.
Rett sindroma ārstēšana
Ārstēšana jāveic pediatram līdz bērna 18 gadu vecumam, un pēc tam seko ģimenes ārsts vai neirologs.
Konsultācijām jānotiek ik pēc 6 mēnešiem, un tajās var novērot dzīvībai svarīgās pazīmes, augumu, svaru, zāļu pareizību, bērna attīstības novērtēšanu, ādas izmaiņas, piemēram, dekubīta brūču klātbūtni, kas ir izgulējumi, kas var inficēties. nāves risks. Citi aspekti, kas var būt svarīgi, ir attīstības un elpošanas un asinsrites sistēmas novērtēšana.
Fizioterapija jāveic visā cilvēka ar Reta sindromu dzīves laikā, un tā ir noderīga tonusa, stājas, elpošanas uzlabošanai, un tādas metodes kā Bobath var izmantot, lai palīdzētu bērnam attīstīties.
Psihomotorās stimulācijas sesijas var notikt apmēram 3 reizes nedēļā, un tās var palīdzēt, piemēram, ar motoru attīstīties, samazināt skoliozes smagumu, kontrolēt droolēšanu un sociālo mijiedarbību. Terapeits var norādīt dažus vingrinājumus, kurus vecāki var veikt mājās, lai katru dienu veiktu neiroloģisko un motorisko stimulu.
Cilvēka ar Reta sindromu mājās ir nogurdinošs un grūts uzdevums. Vecāki var kļūt ļoti emocionāli iztukšoti, tāpēc viņiem var ieteikt sekot psihologiem, kuri var palīdzēt tikt galā ar savām emocijām.
Skatīt Vairāk

5 krūšu masāžas priekšrocības
